Genotype quality control with plinkQC
Hannah Meyer and Maha Syed
2026-03-26
Source:vignettes/plinkQC.Rmd
plinkQC.RmdIntroduction
Genotyping arrays enable the direct measurement of an individual’s genotype at thousands of markers. Subsequent analyses such as genome-wide association studies rely on the high quality of these marker genotypes.
plinkQC facilitates such genotype quality control, including a pre-trained ancestry classifier and relatedness filter optimized to retain the maximally unrelated sample set with highest quality.
plinkQC assumes the genotypes have already been determined from the original probe intensity data of the genotype array and are available in plink format. It wraps around PLINK [2] basic statistics (e.g. missing genotyping rates per individual, allele frequencies per genetic marker) and relationship functions. plinkQC then generates a per-individual and per-marker quality control report and individuals and markers that fail the quality control can subsequently be removed with plinkQC to generate a new, clean dataset. plinkQC can also generate MultiQC compatible reports.
The majority of functions in plinkQC depend on PLINK (version 1.9), which has to be manually installed prior to the usage of plinkQC. The ancestry functions depend on the newer version of PLINK 2.0 (version 2.0).
The protocol can be run via three main functions, the per-individual
quality control (perIndividualQC), the per-marker quality
control (perMarkerQC) and the generation of the new,
quality control dataset (cleanData):
Per-individual quality control
The per-individual quality control with perIndividualQC
wraps around these functions:
check_sex: for the identification of individuals with discordant sex information,check_heterozygosity_and_missingness: for the identification of individuals with outlying missing genotype and/or heterozygosity rates,check_relatedness: for the identification of related individualsancestry_prediction: for prediction of genomic ancestry
Per-marker quality control
The per-marker quality control with perMarkerQC wraps
around these functions:
check_snp_missingnes: for the identifying markers with excessive missing genotype rates,check_hwe: for the identifying markers showing a significant deviation from Hardy-Weinberg equilibrium (HWE),check_maf: for the removal of markers with low minor allele frequency (MAF).
Workflow
In the following, genotype quality control with plinkQC is
applied on a small example dataset with 200 individuals and 10,000
markers (provided within this package). The quality control is
demonstrated in three easy steps, per-individual and per-marker quality
control followed by the generation of the new dataset. In addition, the
functionality of each of the functions underlying
perMarkerQC and perIndividualQC is
demonstrated at the end of this vignette.
To run the ancestry functionality of the package, Plink v2 is needed. Before use, the study data should be in the new hg38 annotation. USCS’s liftOver tool may be needed to map variants from one annotation to another. More details on how to use the tool can be found on the processing HapMap III reference data vignette. We provide an example dataset that is in the hg38 annotation.
Additional loading matrices are needed for the PCA projection used in the model. This is hosted on the plinkQC github repo under the inst/extdata folder located here. Alternatively, the whole github repo can downloaded with
The name of the files (before the .acount or .eigenvec.allele) for
the loading matrices must be included in the path2load_mat
variable.
package.dir <- find.package('plinkQC')
indir <- file.path(package.dir, 'extdata')
qcdir <- tempdir()
name <- 'data.hg38'
path2plink <- "/path/to/plink"
path2plink2 <- "/path/to/plink2"
path2load_mat <- "path/to/load_mat/merged_chrs.postQC.train.pca"Per-individual quality control
For perIndividualQC, one simply specifies the directory
where the data is stored (qcdir) and the prefix of the plink files
(i.e. prefix.bim, prefix.bed, prefix.fam). Per default, all quality
control checks will be conducted.
In addition to running each check, perIndividualQC
writes a list of all fail individual IDs to the qcdir. These IDs will be
removed in the computation of the perMarkerQC. If the list
is not present, perMarkerQC will send a message about
conducting the quality control on the entire dataset.
The ancestry portion of the package requires the variant identifiers
should be formatted similar to the following example: 1:12345[hg38].
perIndividualQC will run
rename_variant_identifiers() to rename the variant
identifiers.
NB: There are limitations on CRAN file sizes. To reduce the
data size of the example data in plinkQC, data.genome has
already been reduced to the individuals that are related. Thus the
relatedness plots in C only counts for related individuals only. For
ancestry identification, the example dataset is very small (10k
genotypes) and contains markers and individuals failing qc controls to
showcase the plinkQC’s functionality. In practice this means
that the overlap in genotypes between the study and reference data is
only 597 SNPs (roughly 0.2%) of the reference dataset. Thus, additional
noise is expected and visible in the ancestry plot below.
perIndividualQC displays the results of the quality
control steps in a multi-panel plot.
fail_individuals <- perIndividualQC(indir=indir, qcdir=qcdir, name=name,
path2plink=path2plink,
path2load_mat = path2load_mat,
path2plink2=path2plink2,
excludeAncestry = c("Africa"),
interactive=TRUE, verbose=TRUE)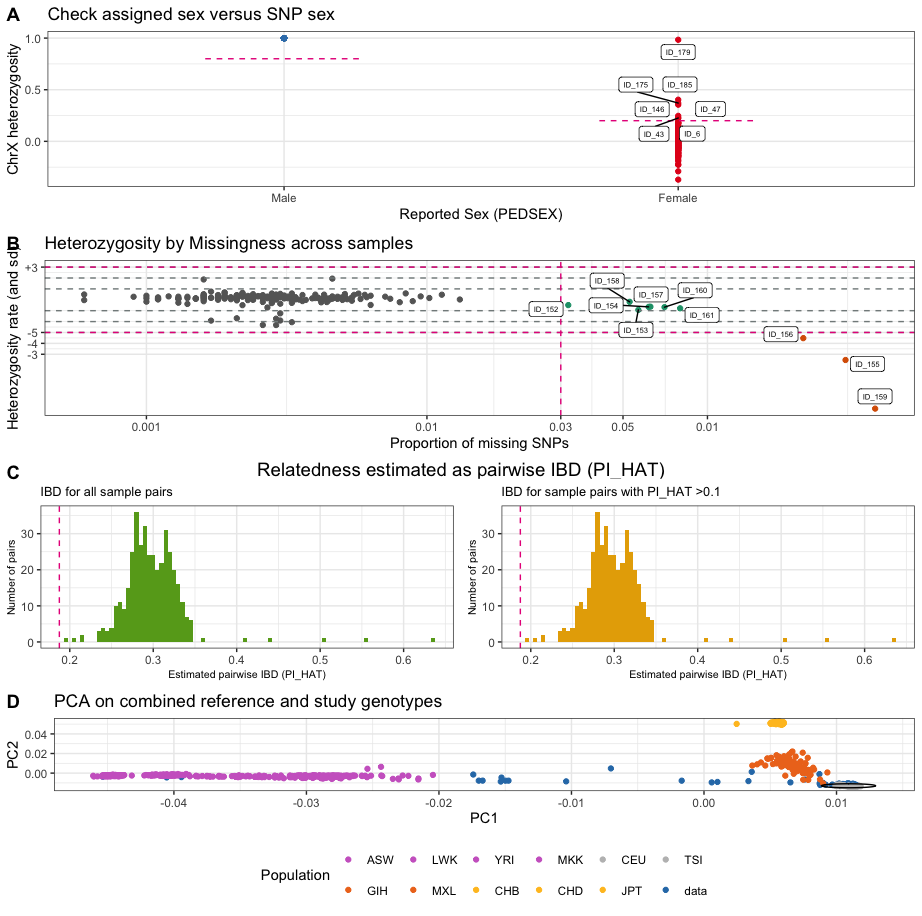
overviewperIndividualQC depicts overview plots of
quality control failures and the intersection of quality control
failures.
overview_individuals <- overviewPerIndividualQC(fail_individuals,
interactive=TRUE)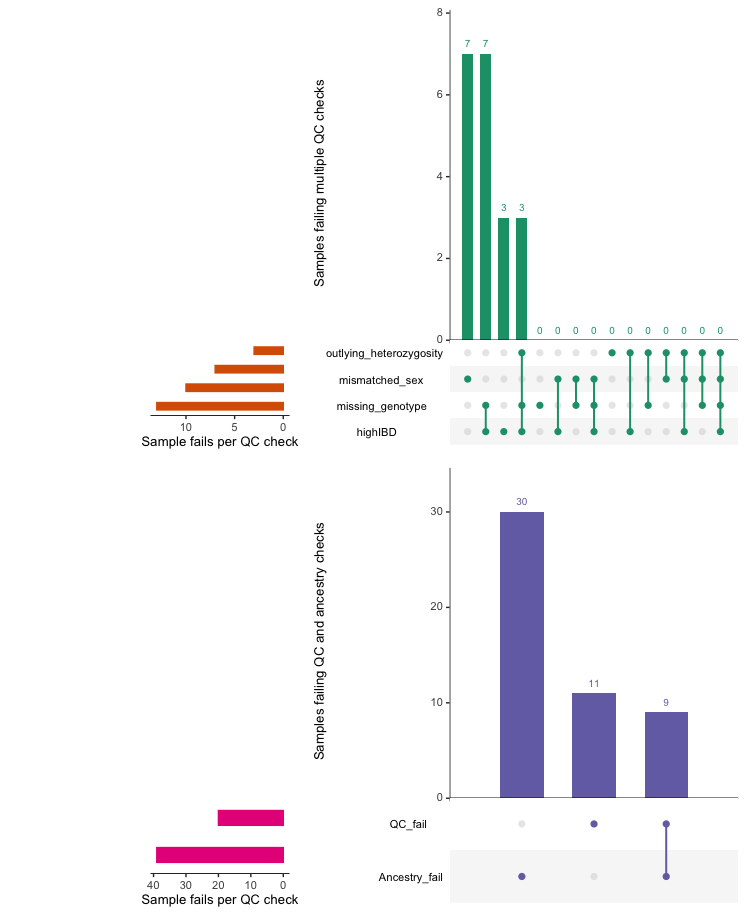
Per-marker quality control
perMarkerQC applies its checks to data in the specified
directory (qcdir), starting with the specified prefix of the plink files
(i.e. prefix.bim, prefix.bed, prefix.fam). Optionally, the user can
specify different thresholds for the quality control checks and which
check to conduct. Per default, all quality control checks will be
conducted. perMarkerQC displays the results of the QC step
in a multi-panel plot.
fail_markers <- perMarkerQC(indir=indir, qcdir=qcdir, name=name,
path2plink=path2plink,
verbose=TRUE, interactive=TRUE,
showPlinkOutput=FALSE)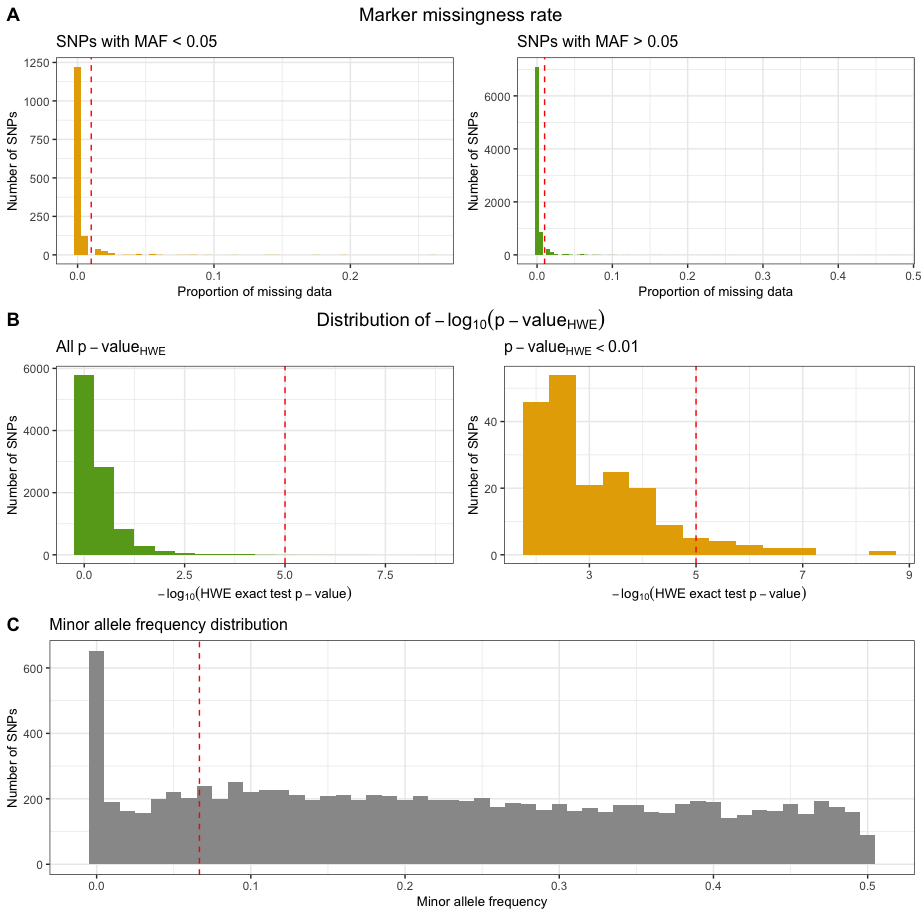
overviewPerMarkerQC depicts an overview of the marker
quality control failures and their overlaps.
overview_marker <- overviewPerMarkerQC(fail_markers, interactive=TRUE)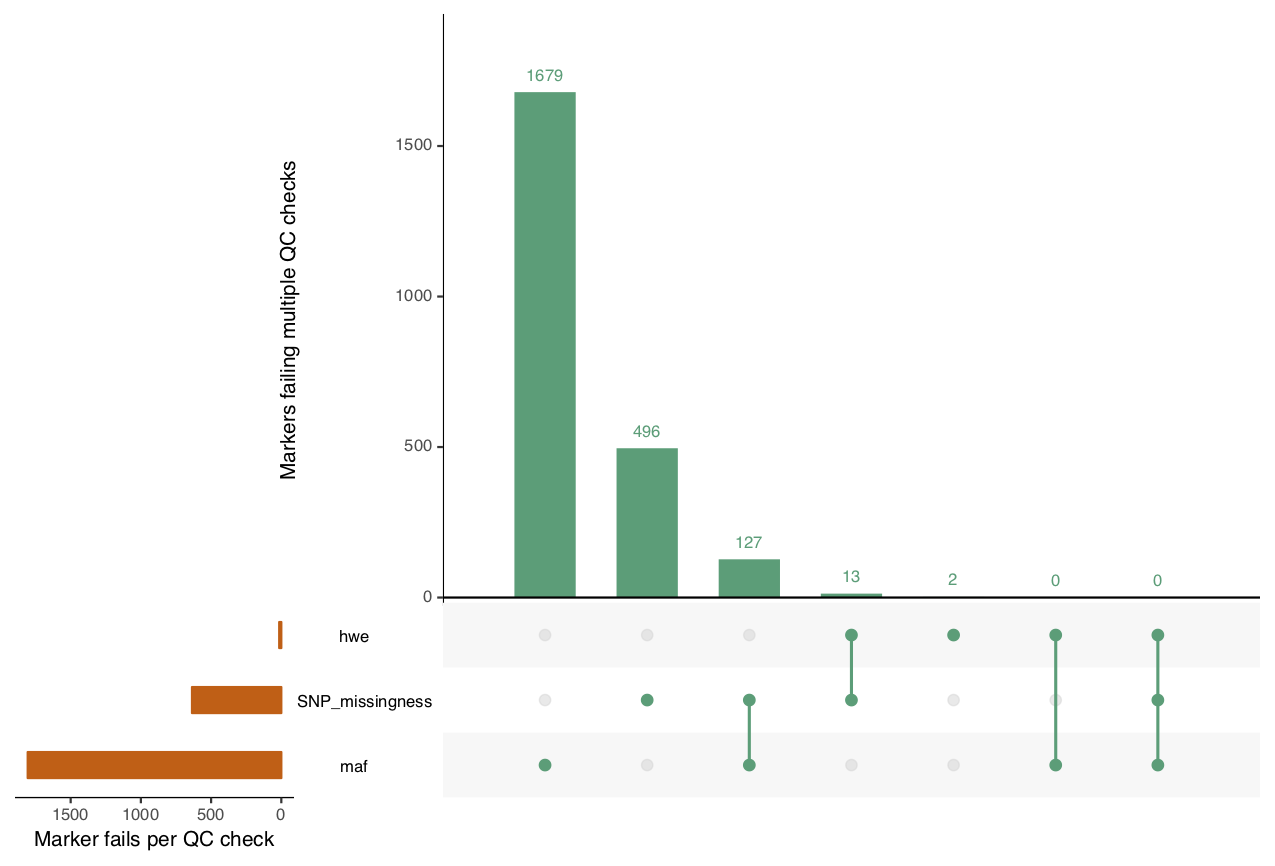
Create QC-ed dataset
After checking results of the per-individual and per-marker quality
control, individuals and markers that fail the chosen criteria can
automatically be removed from the dataset with cleanData,
resulting in the new dataset qcdir/data.clean.bed,qcdir/data.clean.bim,
qcdir/data.clean.fam. For convenience, cleanData returns a
list of all individuals in the study split into keep and remove
individuals.
Ids <- cleanData(indir=indir, qcdir=qcdir, name=name, path2plink=path2plink,
verbose=TRUE, showPlinkOutput=FALSE)Step-by-step
Individuals with discordant sex information
The identification of individuals with discordant sex information
helps to detect sample mix-ups and samples with very poor genotyping
rates. For each sample, the homozygosity rates across all X-chromosomal
genetic markers are computed and compared with the expected rates
(typically
for females and
for males). For samples where the assigned sex (PEDSEX in the .fam file)
contradicts the sex inferred from the homozygosity rates (SNPSEX), it
should be checked that the sex was correctly recorded (genotyping often
occurs at different locations as phenotyping and misrecording might
occur). Samples with discordant sex information that is not accounted
for should be removed from the study. Identifying individuals with
discordant sex information is implemented in check_sex. It
finds individuals whose SNPSEX != PEDSEX. Optionally, an extra
data.frame with sample IDs and sex can be provided to double check if
external and PEDSEX data (often processed at different centers) match.
If a mismatch between PEDSEX and SNPSEX was detected, by SNPSEX == Sex,
PEDSEX of these individuals can optionally be updated.
check_sex depicts the X-chromosomal heterozygosity (SNPSEX)
of the samples split by their (PEDSEX).
fail_sex <- check_sex(indir=indir, qcdir=qcdir, name=name, interactive=TRUE,
verbose=TRUE, path2plink=path2plink)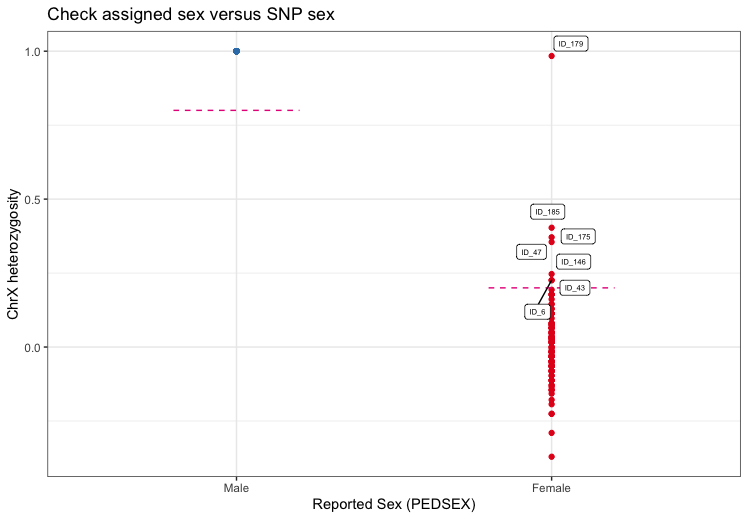
Individuals with outlying missing genotype and/or heterozygosity rates
The identification of individuals with outlying missing genotype
and/or heterozygosity rates helps to detect samples with poor DNA
quality and/or concentration that should be excluded from the study.
Typically, individuals with more than 3-7% of their genotype calls
missing are removed. Outlying heterozygosity rates are judged relative
to the overall heterozygosity rates in the study, and individuals whose
rates are more than a few standard deviations (sd) from the mean
heterozygosity rate are removed. A typical quality control for outlying
heterozygosity rates would remove individuals who are three sd away from
the mean rate. Identifying related individuals with outlying missing
genotype and/or heterozygosity rates is implemented in
check_het_and_miss. It finds individuals that have
genotyping and heterozygosity rates that fail the set thresholds and
depicts the results as a scatter plot with the samples’ missingness
rates on x-axis and their heterozygosity rates on the y-axis.
fail_het_imiss <- check_het_and_miss(indir=indir, qcdir=qcdir, name=name,
interactive=TRUE, path2plink=path2plink)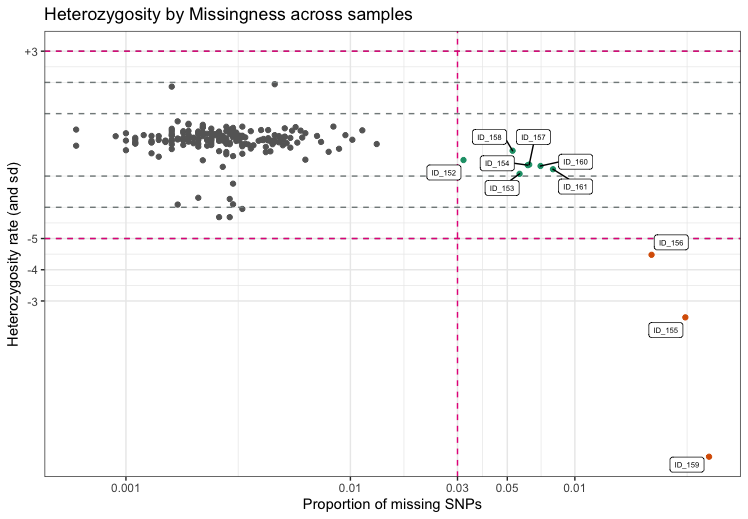
Related individualis
Depending on downstream analyses, it might be required to remove
related individuals from the study. Related individuals can be
identified by their proportion of shared alleles at the genotyped
markers (identity by descend, IBD). Standardly, individuals with
second-degree relatedness or higher will be excluded. Identifying
related individuals is implemented in check_relatedness. It
finds pairs of samples whose proportion of IBD is larger than the
specified highIBDTh. Subsequently, for pairs of individual
that do not have additional relatives in the dataset, the individual
with the greater genotype missingness rate is selected and returned as
the individual failing the relatedness check. For more complex family
structures, the unrelated individuals per family are selected (e.g. in a
parents-offspring trio, the offspring will be marked as fail, while the
parents will be kept in the analysis).
NB: To reduce the data size of the example data in
plinkQC, data.genome has already been reduced to the
individuals that are related. Thus the relatedness plots in C only show
counts for related individuals only.
exclude_relatedness <- check_relatedness(indir=indir, qcdir=qcdir, name=name,
interactive=TRUE,
path2plink=path2plink)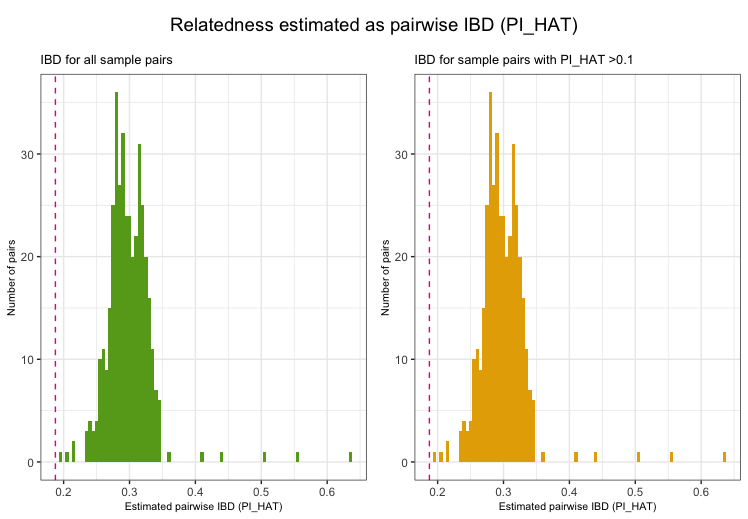
Ancestry Predictions of Data
For ancestry estimation, accessory functions from Plink v2 are needed, as are pre-computed loading matrices for the PCA projection used in the model. These are hosted on the plinkQC github repo under the inst/extdata folder located here. Alternatively, the whole github repo can downloaded with
For this, it is important that the data is in hg38 annotation. USCS’s liftOver tool may be needed to map variants from one annotation to another. More details on how to use the tool can be found on the processing HapMap III reference data vignette.
Additionally, the ancestry prediction function requires the data
variant identifiers in the format: 1:12345[hg38]. The function
ancestry_prediction will call helper functions
rename_variant_identifiers() to format the data as directed
by the parameters. If the data is plink format 2.0, the parameter
plink2format should be set to TRUE.
The function returns: (i) prediction_prob: Dataframe of family ids, sample ids, and model probability of the each ancestry. (ii) prediction_majority: Dataframe of family ids, sample ids, and ancestry with highest model probability. (iii) exclude_ancestry: A list of ids to be excluded based on user-specified ancestries to the ancestral predictions of the samples (iv) p_ancestry: Bar graph of the ancestry model probabilities.
path2plink2 <- "/Users/syed/bin/plink2"
path2load_mat <- "path/to/load_mat/merged_chrs.postQC.train.pca"
anc_check <- ancestry_prediction(indir=indir, qcdir=qcdir, name=name,
interactive=TRUE,
path2plink2=path2plink2,
path2load_mat = path2load_mat,
plink2format = FALSE)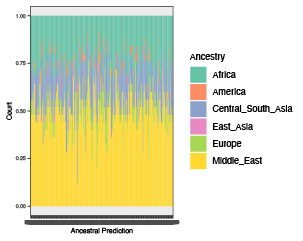
Markers with excessive missingness rate
Markers with excessive missingness rate are removed as they are
considered unreliable. Typically, thresholds for marker exclusion based
on missingness range from 1%-5%. Identifying markers with high
missingness rates is implemented in snp_missingness. It
calculates the rates of missing genotype calls and frequency for all
variants in the individuals that passed the
perIndividualQC.
fail_snpmissing <- check_snp_missingness(indir=indir, qcdir=qcdir, name=name,
interactive=TRUE,
path2plink=path2plink,
showPlinkOutput=FALSE)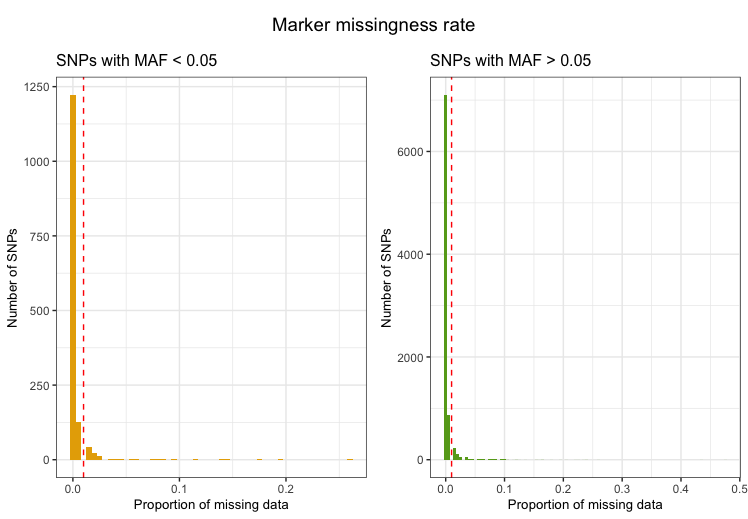
Markers with deviation from HWE
Markers with strong deviation from HWE might be indicative of
genotyping or genotype-calling errors. As serious genotyping errors
often yield very low p-values (in the order of
),
it is recommended to choose a reasonably low threshold to avoid
filtering too many variants (that might have slight, non-critical
deviations). Identifying markers with deviation from HWE is implemented
in check_hwe. It calculates the observed and expected
heterozygote frequencies per SNP in the individuals that passed the
perIndividualQC and computes the deviation of the
frequencies from Hardy-Weinberg equilibrium (HWE) by HWE exact test.
fail_hwe <- check_hwe(indir=indir, qcdir=qcdir, name=name, interactive=TRUE,
path2plink=path2plink, showPlinkOutput=FALSE)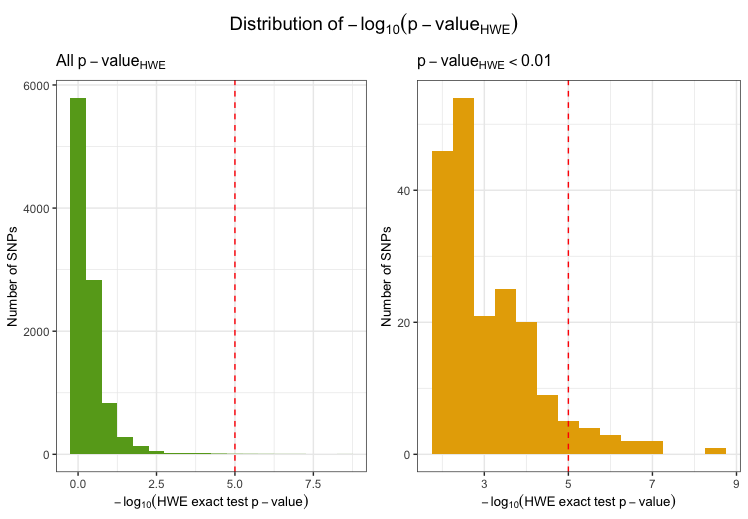
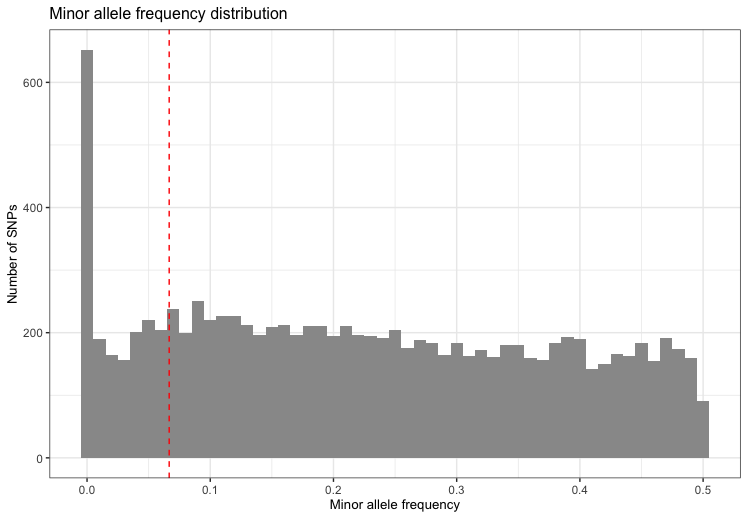
Markers with low minor allele frequency
Markers with low minor allele count are often removed as the actual
genotype calling (via the calling algorithm) is very difficult due to
the small sizes of the heterozygote and rare-homozygote clusters.
Identifying markers with low minor allele count is implemented in
check_maf. It calculates the minor allele frequencies for
all variants in the individuals that passed the
perIndividualQC.
fail_maf <- check_maf(indir=indir, qcdir=qcdir, name=name, interactive=TRUE,
path2plink=path2plink, showPlinkOutput=FALSE)